
本文重點
- 在美國,再生醫療技術需由FDA監管,因為細胞和組織等生物材料可能對患者造成風險,包括污染、免疫排斥和安全顧慮。
- 根據FDA的規定,用於治療、預防或診斷疾病的細胞治療產品都需要進行評估和批准,並需符合優良製造慣例(GMP)的要求。
- 美國將細胞治療分為PHS 361和PHS 351兩大類產品進行管理。其中,符合PHS 361標準的同源使用產品和最小操作產品不需獲得FDA許可或進行臨床試驗。
- 美國在建立較寬鬆的上市途徑的同時,仍需符合FDA的其他要求,如報告任何與產品相關的不良事件和確保製造過程符合優良製造準則等。此外,若製造商聲稱其產品可治療或預防疾病,則須獲得FDA許可,適用於同源使用產品和最小操作產品。
- 歐盟訂有法規命令規範先進醫療技術產品,其中「醫院豁免行為」規定在特定條件下,ATMP無需經歐盟藥物管理局核准,即可在個別歐盟會員國中實施,但生產量或頻率達到一定程度,豁免就會取消。
- 對於我國《再生醫療法》第9條,建議可參考美國的「最小操作」和「同源使用」原則規範風險極小的行為,以及歐盟的醫院豁免制度規範風險大的行為,將「非常規使用」明確列入要件。
- 藥政單位在管制再生醫療產品方面扮演重要角色,因此我國法制宜與國際接軌,以促進產業國際化。
究竟在產業創新、醫療可近性、病人安全保護等面向,如何決定平衡點?再生醫療「技術」不就是一種醫療技術,為何需由食藥署管制?本文擬從歐美的管制架構,說明不同再生醫療技術的管轄權歸屬,以期有助爭議解決。
美國法制原則:「最小操作、同源使用」的風險小產品才能免於藥政單位審查
在美國,為何再生醫療技術需由食品藥品監督管理局(FDA)管制呢?因為,再生醫療治療方法會使用細胞和組織等生物材料,對患者可能造成某些風險。這些風險包括:
- 污染:細胞治療中使用的生物材料可能被病毒、細菌或其他微生物污染,可能在患者身上引起感染或其他不良反應。
- 免疫排斥:細胞治療涉及將來自捐贈者的細胞引入患者體內,這可能引發免疫反應並排斥細胞。
- 安全顧慮:細胞治療可能涉及以未知或無法預料的安全風險方式操縱細胞。
基於這些考量,美國將細胞治療歸類為生物製劑(biologic),須符合FDA監管要求。根據FDA規定,所有用於治療、預防或診斷疾病的細胞治療產品,都須在FDA監管下進行評估和批准。
申請FDA批准前,細胞治療製造商須進行臨床試驗,以評估治療的安全性和有效性。FDA規定細胞治療須符合優良製造準則(Good Manufacturing Practice,GMP)的要求,以確保產品的質量、安全性和純度。
 FDA是美國食品與藥品管理的最高執法機關,並要求細胞治療應符合優良製造原則。圖片來源:Shutterstock
FDA是美國食品與藥品管理的最高執法機關,並要求細胞治療應符合優良製造原則。圖片來源:Shutterstock
根據《聯邦法規》(Federal Registration)第21篇1271條(21CFR 1271),FDA對於人體細胞、組織及基於細胞或組織的產品(human cells,tissues,and cellular and tissue-based products,HCT/Ps)進行風險分類,分為PHS 361及PHS 351兩大類產品進行管理。
其中,PHS 361產品是傳染疾病的風險甚小的產品。只需符合下列所有條件,即可免於遵守FDA上市許可之規範:
「同一手術流程」例外性規定(The same surgical procedure exception,SSP Exception):指在某些情況下,將細胞或組織從患者身體中取出,經過最小程度處理後,再將其重新植入患者身體中的醫療行為。因被視為相同的外科手術程序,故不需遵從FDA的嚴格管制程序。
這類醫療行為須符合以下條件:
- 從患者身體中取出的細胞或組織經過「最小程度操作」(minimally manipulated)。最小操作產品是指未改變細胞的原始特性或生物學功能,且未進行任何重大的物理、化學或酵素處理,不會改變其生理學特性;
- 細胞或組織的重新植入必須發生在同一個手術程序;
- 細胞或組織僅能重新植入原始患者的體內(homologous use)。同源性使用產品必須用於與細胞產品相同的器官或組織,並用於支持、修復或替代組織或器官;
- 操作不包含細胞或組織工程技術或其他類型的生物技術,不與其他藥物或醫療器材結合;
- 操作的目的是恢復或增進患者的生理結構或功能,不會對身體產生系統性作用。
換言之,如果細胞或組織的取出和重新植入符合上述條件,且不改變細胞或組織的特性或應用,這些醫療行為便不需獲得FDA許可或進行臨床試驗。不過,若細胞或組織經過更複雜的操作、處理或轉換後再進行植入,則可能需遵從FDA的許可程序。
若產品無法滿足上述任一條件,則屬於風險較高的PHS 351類產品,須經三階段臨床試驗評估產品的安全及效用,並經過上市前審查,也需符合人體細胞組織優良操作規範(Good Tissue Practice,GTP)及優良製造準則(Good Manufacturing Practice,GPM)。此外,不論PHS 361產品或PHS 351產品,均需符合捐贈者合適性(Donor Eligibility)規範。
即使符合同源使用產品和最小操作產品的標準,製造商仍需遵守FDA的其他要求,如報告任何與產品相關的不良事件,確保製造過程符合優良製造準則等。另外,若製造商聲稱其產品可治療或預防疾病,則必須獲得FDA許可,這也適用於同源使用產品和最小操作產品。
我國俗稱「特管辦法」的「特定醫療技術檢查檢驗醫療儀器施行或使用管理辦法」附件三所列療法,就是以美國這種同源使用及最小操作為原則。其中第16條第2項規定:「前項細胞製備場所之設置,應符合中央主管機關公告之人體細胞組織優良操作相關規範。」亦即仍需符合藥政機關公告的GTP(Good Tissue Practice)規定。
Regenerative Sciences案:FDA對「製造細胞」行為有管轄權
再生科學(Regenerative Sciences)是一家位於美國科羅拉多州的公司,在2012年與FDA爭議「Regenexx細胞治療」是否屬FDA管轄範圍。
本案爭議焦點的再生醫療技術稱為「Regenexx-C」程序,是再生科學公司開發,作為治療關節炎和其他骨科疾病的一種方法。
該製程是先從患者骨髓中提取幹細胞(MSCs),在實驗室中與患者的血液和其他介質混合培養一段時間後,再注射到患者的受傷部位進行治療
該程序包括從患者骨髓或滑膜液樣本提取間質幹細胞,然後在患者自身的血小板溶解液中進行培養增殖;注射到同一患者的骨科損傷部位前,與抗生素混合。
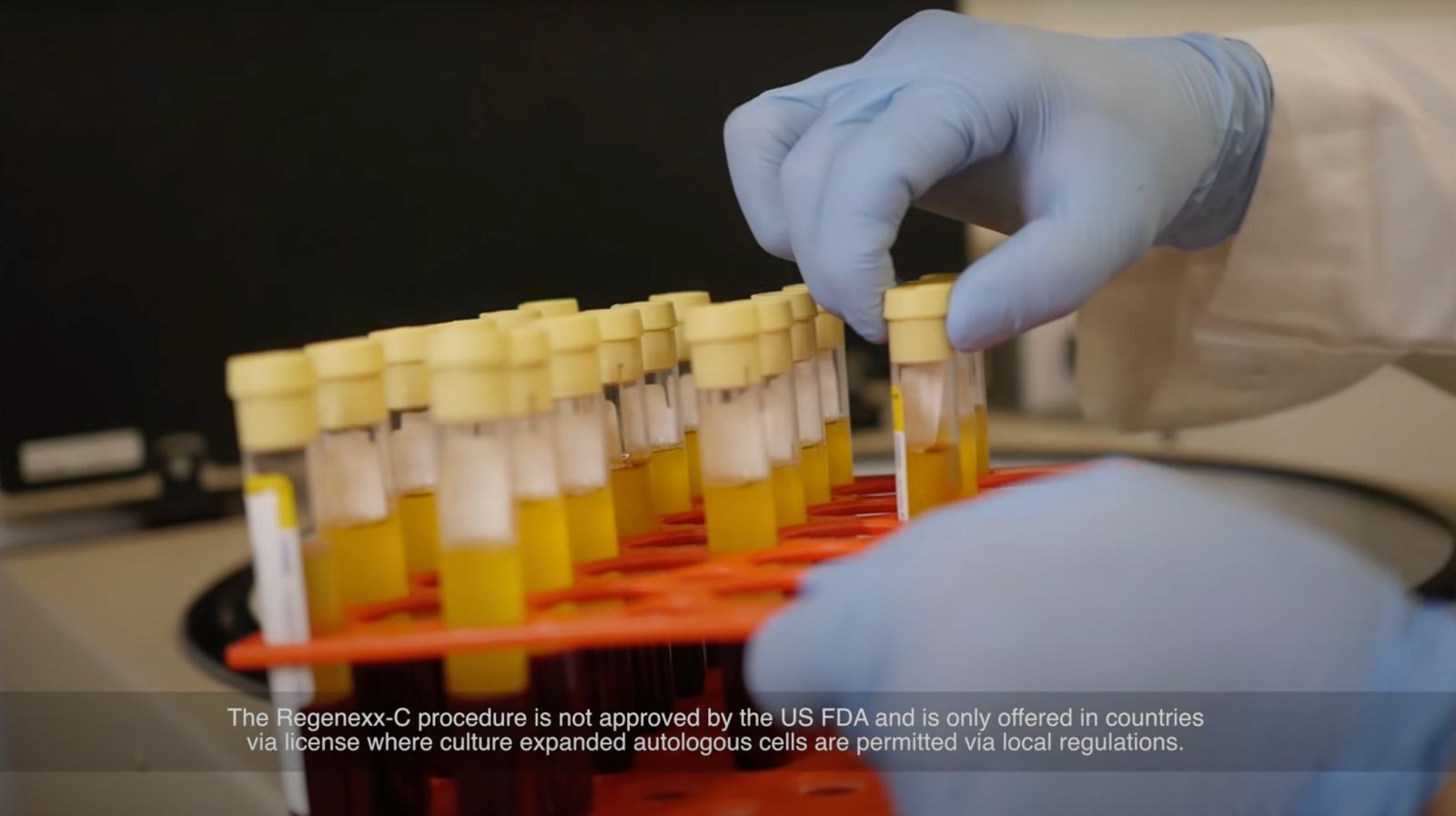 生物學家提取患者幹細胞後,在實驗室培養、增殖患者血液,使細胞增生成原本的100至1,000倍。圖片來源:截自Regenexx影片
生物學家提取患者幹細胞後,在實驗室培養、增殖患者血液,使細胞增生成原本的100至1,000倍。圖片來源:截自Regenexx影片
FDA多次對再生科學未經核准就私自販售產品的行為發出警告,該公司因此提起訴訟,主張Regenexx只是「醫療行為」,FDA無權進行管制,但法院判決再生科學敗訴。
法院認為,Regenexx對幹細胞的操作不僅僅是最小程度操作——在溶解液中培養「增殖」,並「與抗生素混合」等行為,構成生物藥的「製造」(manufacturing)行為,因此FDA有權控制藥品的有效性和製程;即使這些藥品需由醫生開處方,而管制可能會影響醫生的醫療行為。
美國也遇到產業界希望加速產品上市的呼聲,但解決的方式是請藥政機關設計新的上市途徑,而非棄守另建一套醫療體系的管制架構。
由於美國產業界希望放寬細胞治療的管制,且美國病人也推動「嘗試權」(Right to Try,生命危急病人對只完成第一期臨床試驗藥品的近用權)運動,希望讓病人更早使用再生醫療。因此2016年美國國會通過了《21世紀醫療法》(21st Century Cures Act),要求FDA設計出加速再生醫療產品的上市程序。
2017年,FDA因此推出了新的細胞治療框架,稱為「再生醫學先進療法」(Regenerative Medicine Advanced Therapy,RMAT)程序,加速有潛力的細胞治療的發展和批准。符合RMAT要求的細胞治療產品,可獲得加速批准和更快上市時間。
綜合以上,美國的作法,是從FDA方面,建立較寬鬆的上市途徑;而非為了振興產業,就另建一套不同於食藥署的管轄機構。而能否免為FDA管轄,「最小操作」「同源使用」是關鍵詞。(延伸閱讀|跨越死亡之谷:智慧醫材加速的六個法律挑戰)
歐盟:風險高的「醫院豁免行為」僅限於少量生產
在歐洲國家,細胞治療材料的生產質量,是透過指導方針和監管機構的結合進行監管。其中一個關鍵的指導方針是「藥品檢查協作計劃」(PIC/S),這是國際監管機構之間的合作。雖然PIC/S並非歐洲特定的監管機構,但它在調和其成員國,包括許多歐洲國家的「優良製造範例」(GMP)標準方面,發揮至關重要的作用。
歐盟對於以產品上市為目的或預備以工業或包含工業程序之方式生產的先進醫療技術(下稱ATMPs),訂有相關法規命令,包括體細胞治療產品(CTMP)、基因治療產品(GTMP)、組織工程產品(TEP)以及合併的先進治療產品。
這些法規要求遵守與一般藥品相同的法規標準,包括優良製造標準、樞紐性臨床試驗及小兒試驗計畫等。由於這些法規加重了研究者及製造商的成本及負擔,因此在一般標準之外,也設立了例外程序,使得在特定條件下,這類ATMP無需經歐盟藥物管理局核准,即可在個別歐盟會員國中實施,此即「醫院豁免行為」(Hospital Exemption)。
「醫院豁免行為」規定在歐盟Article 28 of Regulation No. 1394/2007。該規定允許藥廠在醫院或醫師的醫療處方下,進行少量客製化(non-routine)製造行為;但若生產量或頻率達到一定程度,就可能被認定為以上市為目的,此時豁免就會取消。因此,製造商須向藥政機關通報實際製造頻率,亦即藥政機關對細胞製造行為仍有管轄權,只是在這些少量個案不要求上市許可。
這個規定並非提供藥廠另一種上市途徑,而是考慮到少數個別病患,提供比「恩慈療法」更寬鬆的條件。此舉不需病人陷於生命危險,或市場上不存在其他有效治療藥物的前提,且可以在沒有臨床試驗的情況下單獨存在。具體而言包括下列條件:
- 產品按照會員國制定的特定質量標準,在醫院內部的藥房或其他授權場所製備;
- 產品僅在醫院內、且由直接負責使用的醫師專責使用,該醫師對其使用負有個人和直接責任;
- 產品以非常規方式(non-routine basis)提供給個別患者使用;
- 產品的起始材料已通過採購、檢查和加工以確保最終產品的安全性、質量和功效;
- 產品的製備方式不會對產品的質量、安全性和功效產生負面影響;
- 產品符合會員國制定的特定質量標準。
在醫院豁免情況下,PIC/S、GMP和GCP的具體要求,可能不像中央化市場授權的ATMP那樣直接適用,但仍然受到一定品質和安全要求的限制,包括可追溯性和藥物監測。使用醫院豁免的ATMP的醫院和醫療從業人員,須確保從供體到受體之間的產品可追溯性,並建立報告和處理任何與ATMP使用相關的不良反應的系統,以確保接受這些治療的患者的福祉。
 歐盟以PIC/S等指導標準,監管國際間的細胞治療材料合作。圖片來源:Shutterstock
歐盟以PIC/S等指導標準,監管國際間的細胞治療材料合作。圖片來源:Shutterstock
台灣《再生醫療法》如何調整才能接軌國際?
台灣《再生醫療法》第9條是否會依既有草案通過?如果要調整,可朝何種方向進行?從以上歐美經驗看來,我們可針對有爭議的第2款和第3款,提出以下建議:
- 第3款「提供不含基因改造或轉殖之人類細胞及其衍生物之細胞治療」,是指風險極小的行為,可朝美國的「最小操作」和「同源使用」原則來規範。
- 第2款「經執行人體試驗結果,證實其安全性及初步療效」,是針對風險大的行為,包括異體(甚至異種)及超過最小操作的行為。此類型可參考歐盟的醫院豁免制度,將「非常規使用」明確列入要件。
目前對該款的說法中,雖有提到「若數量大就應改走再生醫療製劑的上市途徑」;但從該款草案的構成要件看來,實在找不到「量大就要改走再生醫療製劑途徑」的說法。歐盟各主要國家對於何謂「非常規使用」均有頒布指引,作更具體的定義(例如一年10件);將「非常規」納入要件並明確定義,或許是此案可行的方向。
再生醫療涉及創新技術及個人化需求,全盤照用既有的生物藥上市途徑有困難,故各國均試圖在「病人安全」及「醫療近用權」之間取得平衡;但原則上,關於「個人化產品」的主要管制創新,仍來自於藥政單位。
最大差別在於,藥政單位本質上具有國際法規調和的性質,會跟隨國際趨勢進行法規創新;許多個人化醫材,如假牙或3D生物列印,藥政單位均能設計出特別的簡易上市途徑。對於再生醫療,世界許多先進國家亦由藥政單位扮演管制主力,並透過法制創新,加速再生醫療的上市,以及降低上市成本。
就一項新興技術的推廣而言,最重要的是建立國人對此技術的信任。走得快不如走得穩。我國法制宜與國際接軌,建立產業國際化的思維,以強化民眾對再生醫療產業的信心。
關於智慧醫療,你可能也有興趣:









